Institutional Ethics Commitee (IEC)
IEC-GDCHN operates to ensure the protection of the rights and welfare of human participants in biomedical experimental and behavioural research conducted in GDCH, Nagpur. IEC-GDCHN reviews all types of research proposals involving human participants with the view to safeguard the dignity, rights, safety and wellbeing of all actual and potential research participants before approving the research proposals. The goals of research, however important, should never be permitted to override the health and wellbeing of research subjects.
IEC-GDCHN was established in the year 2014 and since then regular meetings are being conducted to review scientific proposals of under-graduate, post-graduate, fellowship and PhD students as well as proposals from faculty. The review process is as follows-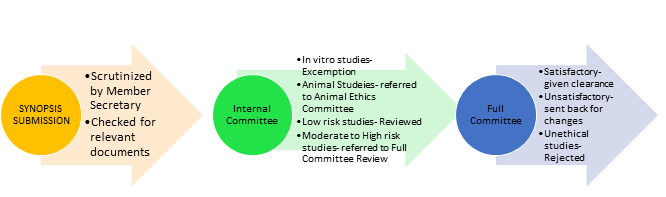
- Meetings are planned according to the workload.
- Independent experts/consultants to offer their expert opinions are invited as per need.
- The entire functioning of the meeting is documented and signature of Chairman is taken after every meeting.
GOVT. DENTAL COLLEGE & HOSPITAL, NAGPUR.
INSTITUTIONAL ETHICS COMMITTEE GOVERNMENT DENTAL COLLEGE AND HOSPITAL, NAGPUR
(IEC—GDCHN)
STANDARD OPERATING PROCEDURES (SOPs)
Dr Darshan Dakshindas
STANDARD OPERATING PROCEDURES (SOPs) FOR INSTITUTIONAL ETHICS COMMITTEE (IEC) GOVERNMENT DENTAL COLLEGE & HOSPITAL, NAGPUR (IEC-GDCHN)
EFFECTIVE DATE: 01.01.2021 NEXT REVIEW DATE: 31.12.2024
The location and business address of the committee: Institutional Ethics Committee, GDCH, Nagpur
Room No-20 Ground Floor
Government Dental College & Hospital, GMC Campus, Medical Square, Nagpur-Maharashtra, India Pin- 440003
Phone No- 0712-2744496
Standard Operating Procedures for Institutional Ethics Committee, GDCHN (IEC GDCHN) is a content of Compendium on scientific research and publications at GDCH, Nagpur.
- Standard Operating Procedures (SOPs) ICH-GCP guideline defines SOPs as “Detailed, written instructions to achieve uniformity of the performance of a specific function”. SOPs provide the essential link between the guideline on one hand and the actual practice on the other. It is but natural therefore that, all the individual participants, performing their primarily required duty or a specific job and belonging to each and every category of the stakeholder has not only to have a well-defined SOPs but also to observe/follow it very carefully. It goes without saying, therefore, that each stakeholder has to have separate SOPs and IEC is no exception to the rule.
SOPs is for the methodical functioning of any important work to be undertaken, a proper, stepwise, work procedure is necessary. In general, in any SOPs the steps given should be reproducible, e.g. in the case of clinical trials, it will be neither proper nor acceptable to have SOPs that can be applied to just one or specific clinical trial. Broadly speaking SOPs can be in four different areas covering (i) organization of study in general, (ii) prior to study, (iii) Actual or during, and (iv) End of the study.
The SOPs should have the following general outline:
- Must have a number with title or checklist. A set SOPs need not have a checklist, but if included it should be sub- numbered, incorporating the corresponding SOPs
- Reference, if any, to other related
- Person/Personnel i.e. who carried out / who all carry out the
- When and how the procedure is carried out (Note: These are especially required for other stakeholders in clinical trials, as compared to the )
- Date of version in use; date of revision/ amendment/ replacement/ automatically replacing the previous version / name of the person or body responsible for the
- The process of review and revision of SOPs should usually be a regularly done exercise, say every few months. A team should preferably do it. The old version should be archived after approval and implementation of the revised version. This is necessary because Regulatory Authorities may want to know some relevant answer to the question that might
Some benefits of SOPs are:
- It provides a written record of the process. Processes used by several individuals are applied (more) consistently. Team member confidence is increased and performance is enhanced. It helps with the training of new
- Reduces supervisory time/effort. However, these can also be generally applicable to other
- As far as this IEC-GDCHN related Work Document is concerned, a properly constituted IEC, functioning regularly and following its own Standard Operating Procedures is a must. Then alone the studies approved by the IEC stand a good chance of the global acceptability of the outcome of their quality
The objective of Standard Operating Procedures (SOPs) is to ensure quality and consistency in review of clinical research proposals and to follow the ICMR and national ethical guidelines for biomedical research on human subjects.
- Composition of Institutional Ethics Committee
ECs should be multi-disciplinary and multi-sectoral. There should be adequate representation of age and gender.
Preferably 50% of the members should be non-affiliated or from outside the institution. The number of members in an EC should preferably be between 07 and 15 and a minimum of five members should be present to meet the QUORUM requirements. The EC should have a balance between medical and non-medical members/technical and non- technical members, depending upon the needs of the institution.
Generally, the representation on the IEC shall be:
- Chairperson (outside Institution-Nominated by the Dean)
- Member Secretary from Institute
- One basic medical scientist (preferably one Pharmacologist)
- One Clinician
- One legal expert or retired Judge
- One social scientist/ representative of non-governmental organization/ Philosopher/ Ethicist/ Theologian or a similar person
- One lay person from the
- Chairperson-(Non-affiliated)-A well-respected person from any background with prior experience of having served/ serving in an
- Member Secretary-(Affiliated)-
- Should be a staff member of the institution
- Should have knowledge and experience in clinical research and ethics and be motivated and have good communication skills
- Should be able to devote adequate time to this activity which should be protected by the institution
- Basic Medical Scientist(s)- (Affiliated/ Non-Affiliated)-
- In case of EC reviewing clinical trials with drugs, the basic medical scientist should preferably be a pharmacologist
- Clinician(s)- (Affiliated/ Non-Affiliated)
- Should be individual/s with recognized medical qualification, expertise and training
- Legal expert/s- (Affiliated/ Non-Affiliated)-
- Should have a basic degree in Law from a recognized university, with experience
- Desirable: Training in medical
- Social scientist/ philosopher/ ethicist/theologian (Affiliated/ Non-Affiliated)-
- Should be an individual with social/behavioural science/ philosophy/ religious qualification and training and/or expertise and be sensitive to local cultural and moral values. Can be from an NGO involved in health-related activities
- Lay person(s)- (Non-affiliated)
- Literate person from the public or community
- Has not pursued a medical science/Health related career in the last 5 years
- May be a representative of the community from which the participants are to be drawn
- Is aware of the local language, cultural and moral values of the community
- Desirable: involved in social and community welfare activities
-Member/s should be sufficiently qualified through the experience and expertise and sensitive to such issues as community attitudes, to promote respect for its advice and counsel in safeguarding the rights and welfare of human subjects.
-In addition to possessing the professional competence necessary to review the specific research activities, the member/s should be able to ascertain the acceptability of proposed research in terms of institutional commitments and regulations, applicable law, and standards or professional conduct and practice. (Ref 21 CFR part 56.107)
-A member of IEC can be a part of any other IRB/IEC
- Tenure: Membership Duration
- The tenure for Members of the IEC is for a period of Three (03) Years.
- It can be extended to another three (03)
- There will be no bar on the members serving for more than one term but it is desirable to have approximately one third fresh
- A member can be replaced in the event of long-term non- availability (three consecutive meetings). Authority to replace the member shall remain with the Chairman
- Members should maintain confidentiality of all discussions during the meeting and sign a confidentiality form at the start of their term. Each member of the committee will submit a declaration to maintain the confidentiality of the documents submitted to them during their membership period.
- Conflict of interest if any, shall be declared by members of the Institutional Ethical Committee at the beginning of every meeting.
To establish polices for removal or Resignation / Replacement of Members Chairman and Member Secretary are responsible for implementing this SOP.
Term of appointment Members of IEC will be appointed for period of 03 years initially which could be extended for another term of 03 years. Extension of membership will be based on the recommendation of the Chairman & Member Secretary of IEC.
Policy for removal of member
- A member may be relieved or terminated of his/her membership in case of conduct not suitable for a member of the Ethics
- Inability to participate in the meetings on any grounds for more than 3 meetings of
- The membership shall be reviewed by the Registrar & chairman, if the member is a regular
- If deemed necessary, the IEC may decide to terminate the membership and recommend to the Chairman IEC for necessary
- In all such situations/circumstances, member secretary will serve a letter of termination to the
- Documentation of the termination will be recorded in the meeting minutes of the next duly constituted IEC meeting and IEC membership circular/roster will be Resignation / Replacement procedure
The members who have resigned may be replaced at the discretion of the appointing authority for the same.
IEC members who decide to resign must provide the Chairman & Member Secretary of IEC the written notification of their proposed resignation date at least 30 calendar days prior to the next scheduled meeting.
In case of resignation, chairman & member secretary would appoint a new member, falling in the same category of membership ex. NGO representative with NGO representative.
- Members should be trained in human research protection, EC functions and SOPs, and should be conversant with ethical guidelines, GCP guidelines (if applicable) and relevant regulations of the
- EC members should undergo initial and continuing training in human research protection, applicable EC SOPs and related regulatory requirements. All trainings should be documented.
- Any change in the relevant guidelines or regulatory requirements should be brought to the attention of all EC members.
- EC members should be aware of local, social and cultural norms and emerging ethical
- IEC/IRB Functions:
Roles and responsibilities of the EC
- The basic responsibility of an EC is to ensure protection of the dignity, rights, safety and well-being of the research participants.
- The EC must ensure ethical conduct of research by the investigator
- The EC is responsible for declaration of conflicts of interest to the Chairperson, if any, at each meeting and ensuring these are recorded in the
- The EC should perform its function through competent initial and continuing review of all scientific, ethical, medical and social aspects of research proposals received by it in an objective, timely and independent manner by attending meetings, participation in discussion and
- The EC must ensure that universal ethics values and international scientific standards are followed in terms of local community values and
- The EC should assist in the development and education of the research community in the given institute (including researchers, clinicians, students and others), responsive to local healthcare
- Responsibilities of members should be clearly defined. The SOPs should be given to EC members at the time of their appointment.
- The EC should ensure that privacy of the individual and confidentiality of data including the documents of EC meetings is
- The EC reviews progress reports, final reports and AE/SAE and gives needful suggestions regarding care of the participants and risk minimization procedures, if
- The EC should recommend appropriate compensation for research related injury, wherever
- The EC should carry out monitoring visits at study sites as and when
- The EC should participate in continuing education activities in research ethics and get updated on relevant guidelines and
- The EC may see that conduct of same/similar research by different investigators from same institution is ‘Me too’ research (replicative) should not to be encouraged and submission of same research to different funding agencies should not be accepted.
- Roles and Responsibilities of IEC members: Chairperson
- Conduct EC meetings, accountable for independent and efficient functioning of the committee
-Ensure active participation of all members (particularly non- affiliated, non-medical/ non- technical) in all discussions and deliberations
-Ratify minutes of the previous meetings
-In case of anticipated absence of Chairperson at a planned meeting, the Chairperson should nominate a committee member as Acting Chairperson or the members present may elect an Acting Chairperson on the day of the meeting. The Acting Chairperson should be a non-affiliated person and will have all the powers of the Chairperson for that meeting.
-Seek COI declaration from members and ensure quorum and fair decision making.
-Handle complaints against researchers, EC members, conflict of interest issues and requests for use of EC data, etc.
Member Secretary-
- Organize an effective and efficient procedure for receiving, preparing, circulating and maintaining each proposal for review.
- Schedule EC meetings, prepare the agenda and minutes
- Organize EC documentation, communication and archiving
- Ensure training of EC secretariat and EC members
- Ensure SOPs are updated as and when required
- Ensure adherence of EC functioning to the SOPs
- Prepare for and respond to audits and inspections
- Ensure completeness of documentation at the time of receipt and timely inclusion in agenda for EC review.
- Assess the need for expedited review/ exemption from
review or full review.
- Assess the need to obtain prior scientific review, invite independent consultant, patient or community representatives.
- Ensure quorum during the meeting and record discussions
and decisions.
Basic Medical Scientist(s)-
- Scientific and ethical review with special emphasis on the intervention, benefit-risk analysis, research design,
methodology and statistics, continuing review process, SAE, protocol deviation, progress and completion report
- For clinical trials, pharmacologist to review the drug safety and
Clinician(s)-
- Scientific review of protocols including review of the intervention, benefit-risk analysis, research design, methodology, sample size, site of study and statistics
- Ongoing review of the protocol (SAE, protocol deviation or violation, progress and completion report)
- Review medical care, facility and appropriateness of the principal investigator, provision for medical car, management and
- Thorough review of protocol, investigators brochure (if applicable) and all other protocol details and submitted documents.
Legal expert/s-
- Ethical review of the proposal, Informed Consent Document (ICD) along with translations, Memorandum of Understanding (MOU), Clinical Trial Agreement (CTA), regulatory approval, insurance document, other site approvals, researcher’s undertaking, protocol specific other permissions, such as, stem cell committee for stem cell research, Human Mesenchymal stem cells (HMSC) for international collaboration, compliance with guidelines etc.
- Interpret and inform EC members about new regulations if any Social scientist/ philosopher/ ethicist/theologian
- Ethical review of the proposal, ICD along with the translations.
- Assess impact on community involvement, socio–cultural context, religious or philosophical context, if any
- Serve as a patient/participant/ societal /community representative and bring in ethical and societal concerns. Lay person(s)-
- Ethical review of the proposal, ICD along with translation(s).
- Evaluate benefits and risks from the participant’s perspective and opine whether benefits justify the risks.
- Serve as a patient/participant/ community representative and bring in ethical and societal concerns.
- Assess on societal aspects if
- Record Keeping and Archiving:
- Curriculum Vitae (CV) of all members of
- Minutes of all meetings duly signed by the Chairperson. Copies of all correspondence with members, researchers and other regulatory
- Copy of existing relevant national and international guidelines on research ethics and laws along with amendments.
- All study related documents (study protocols with enclosed documents, progress reports, and SAEs.) should be archived for minimum of five years after the completion of study. A copy of filled Clinical Record Form (CRF) shall remain with the PI for minimum of ten
- Final report of the completed
- IEC-Operations
- Meeting: Office and Conduct of the Meeting
- The Chairperson will conduct all meetings of the Institutional Ethics Committee. If for reasons beyond control, the Chairperson is not available, an alternate Chairperson will be elected by the members present from among
- The Member Secretary will be responsible for organizing the meetings, maintaining the records and communicating with all concerned. He/she will prepare minutes of the meetings and get them approved by the Chairperson before communicating to members and Principle
- Chairman & member secretary are responsible for implementing this
- The Member Secretary in consultation with the chairman may convene the IEC meeting once in every
- Additional review meeting can also be held with short notice as and when
- All members will receive notification of meeting schedules in
- A minimum of five persons is required to form the quorum without which a decision regarding the research would not be taken. The quorum would have at least one representative from the following group:
- One basic medical scientist (preferably one Pharmacologist)
- One Clinician
- One legal expert or retired Judge
- One social scientist/ representative of non- governmental organization/ Philosopher/ Ethicist/ Theologian or a similar person
- One lay person from the viii Minutes of the IEC meetings, all the proceeding and deliberation will be documented.
Applicant investigator may be invited to present the proposal or elaborate on specific issue.
The Chairperson will conduct all meetings of the Institutional Ethics Committee. If for reasons beyond control, the Chairperson is not available, an alternate Chairperson will be elected by the members present from among themselves.
- Quorum Requirements
A minimum of 5 members including at least one member from the related speciality, in which presentation is due, should be present. All decisions will be taken in the meetings and not by circulation of project proposals. As per revised “Schedule Y’’ of Drugs & Cosmetics Act, 1940, amended in 2005, the ethics committee approving drug trials will have in the quorum at least one representative from the following groups:
- One basic medical scientist (preferably one pharmacologist).
- One clinician
- One legal expert or
- One social scientist
- One lay person from the community
- Independent Consultants:
Institutional Ethics Committee may call upon subject experts as consultants for review of selected research protocols.
These experts may be specialists in ethical or legal aspects, specific diseases or methodologies, or represent specific communities; patient groups or special interest groups e.g. cancer patients, HIV/AIDS positive persons or ethnic minorities. They will not take part in the decision making process and shall have no voting rights.
- Submission of documents for IEC review: Details of documents to be submitted for EC review
- Covering letter to the Member Secretary
- Type of review requested
- Application form for initial review
- The correct version of the informed consent document (ICD) in English and the local language(s). Translation and back translation certificates (if applicable)
- Case record form/questionnaire
- Recruitment procedures: advertisement, notices (if applicable)
- Patient instruction card, diary, etc. (if applicable)
- Investigator’s brochure (as applicable for drug/biologicals/device trials)
- Details of funding agency/sponsor and fund allocation (if applicable)
- Brief curriculum vitae of all the study researchers
- A statement on COI, if any
- GCP training certificate (preferably within 5 years) of investigators (clinical trials)
- Any other research ethics/other training evidence, if applicable as per EC SOP
- List of ongoing research studies undertaken by the principal investigator (if applicable)
- Undertaking with signatures of investigators
- Regulatory permissions (as applicable)
- Relevant administrative approvals (such as HMSC approval for International trials)
- Institutional Committee for Stem Cell Research (IC-SCR) approval (if applicable)
- MoU in case of studies involving collaboration with other institutions (if applicable)
- Clinical trial agreement between the sponsors, investigator and the head of the institution(s) (if applicable).
- Documentation of clinical trial registration (preferable)
- Insurance policy (it is preferable to have the policy and not only the insurance certificate)for study participants indicating conditions of coverage, date of commencement and date of expiry of coverage of risk (if applicable)
- Indemnity policy, clearly indicating the conditions of coverage, date of commencement and date of expiry of coverage of risk (if applicable)
- Any additional document(s), as required by EC (such as other EC clearances for multicentric studies)
- Protocol
- Submission & Review Procedure
1- Researchers should submit research proposals as soft or hard copies to the Member Secretary for review in the prescribed format and required documents as per EC SOPs.
This list is subject to modifications, depending on the type of research, EC SOPs and institutional policies.
- All relevant documents should be enclosed with application.
- For projects within the GDCH,Nagpur, no fee will be charged.
- For trials from outside agencies/sponsors, fee of Rs 30,000 is to be deposited along with application
- The required number of copies of the proposal (14 for ICMR and outside funded projects and for in-house projects as specified) along with the application and documents in prescribed format duly signed by the PI and Co-investigators/ Collaborators should be forwarded by the Head of the Department (Institute).
- The Member Secretary will acknowledge the receipt of proposal and indicate any lacunae. Missing information should be replied within two weeks or as specified
- The date of meeting will be intimated to the PI who should be available to offer clarifications if
- The decision of Institutional Ethics Committee will be communicated in writing. If revision is to be made, the revised document in required number of copies should be submitted within a stipulated period of time as specified in the
- Review of Projects:
- Meetings of Institutional Ethics Committee shall be held at scheduled intervals as prescribed. Additional meetings will be held as and when
- The proposals will be sent to members at least 2 weeks in advance.
- The Member Secretary shall screen the proposals for their completeness and depending on the risk involved categorize them into three types, namely-
Exemption from review, Expedited review, and Full committee review.
- Decisions will be taken by consensus after discussions, and voting will be done if
- Principle Investigator should be available during the meeting and will give brief presentation of his proposal. He may be asked for any
- Independent consultants/Experts may be invited to offer their opinion on specific research
- The decisions of the meeting shall be recorded in the form of minutes and shall be confirmed during the next meeting.
- Element of Review of Proposed Projects:
- Scientific design and conduct of the
- Approval of scientific review committee/Research committee and other regulatory
- Assessment of predictable risks, harms and potential benefits.
- Procedure for selection of subjects including inclusion/exclusion, withdrawal criteria and other issues like sample size and advertisement
- Management of research related injuries, adverse events and compensation
- Justification for placebo in control arm, if
- Availability of products to the trial subjects after the study, if
- Patient information sheet and informed consent form in English/Hindi and local
- Protection of privacy and confidentiality of
- Involvement of the community, wherever
- Protocol and proforma of the study including the consent form.
- Plans for data analysis and
- Adherence to Regulatory requirements and applicable guidelines.
- Competence of Investigators, research and supporting staff.
- Facilities and Review Process:
- A member shall withdraw from the meeting during the decision procedure concerning an application where a conflict of interest arises. This shall be indicated to the chairperson prior to the review of the application and recorded in the minutes.
- Only members will make the decision. The decisions shall be taken in the absence of Investigators, representatives of sponsors and Decision may be of approval, rejection or recommendation to revise the proposals. Specific suggestions for modifications and reasons for rejection should be
- Revised proposals may be subjected to an expedited review.
- All approved proposals will be subject to the laid down standard conditions. Additional conditions may be added by the Institutional Ethics
- Expedited Review: Proposals which are recommended for minor revisions will be reviewed by a sub committee appointed by the Institutional Ethics Committee (IEC) for clearance and approved by the Chairperson. The approvals will be reported in the next Institutional Ethical Committee meeting by Secretary. The revised form of proposals requiring major changes will be reviewed at the next IEC meeting. Rejected proposals may be reconsidered only if a very strong background is there.
- Periodic updates to be submitted to IEC
- All amendments in protocol/ ICF/ IB or any new information available related to IEC approved protocols must be submitted to
- The investigator/Sponsor is responsible to supply ongoing safety updates, progress reports (periodic) of trial status report, re-approvals (if required) to the IRB/IEC.
- PI is required to inform IEC in writing after the site is initiated
- Half yearly progress report to be submitted in the last week of every December and June. PI’s are requested to submit 2 copies of each report to IEC, one to be given back to PI, one for IEC
- After receiving IEC approval, even if site is not initiated half yearly report must be submitted mentioning reasons for not initiating the site
- The P.I. will be required to intimate the name/s designation and nature of work/ responsibilities entrusted to all the members of his/her team in the said protocol. This information will have to be submitted by the P.I. along with the submission letter addressed to the Member Secretary. Change/s in such a job/responsibility profile will be immediately intimated to the Member
- If an assistant whose name is not intimated is entrusted the said job/ responsibility, it will be considered as a breach and the IEC will take appropriate action in the matter and this decision shall be
- The Principal Investigator and the said team shall abide by privacy and confidentiality when the study is approved by the
- Communication of decision of Institutional Ethics Committee
- Decision will be communicated to Principal Investigator (PI) by the Member Secretary in
- Suggestions for modifications and reasons for rejection shall also be communicated to the
- Reporting of Serious Adverse Event (SAE)
When a subject participating in the study develops any SAE , the PI must promptly report the incident to IEC as well as concerned authorities including regulatory bodies if applicable. Following are the rules at GDCH,Nagpur.
- In the case of site related serious adverse event, the Initial SAE report along with copy of SAE form Appendix XI ( as defined by Schedule Y amendment 30 Jan 2013) is to be submitted within 24
- If the adverse event was anticipated in the protocol and the subject was informed about the possibility of event in the Informed Consent Form (ICF) there is no need to inform IEC unless the adverse event was unexpectedly serious, life threatening or
- If the adverse event was unanticipated, unexpectedly serious, life-threatening or fatal, the adverse event must be reported to IEC through Member Secretary within 24
- If the study is being supported by an industry sponsor, the PI is also responsible for notifying the sponsor. The sponsor must notify the regulatory authorities within 24
- If the PI holds the “Investigational New Drug or Device” in his/her name, he/she is required to notify the regulatory authorities of the adverse event within 24 hours in addition to notifying it to
- Notifying to IEC does not relieve the PI from his/her responsibility to notify the sponsor or regulatory
- The PI must submit a written report of the adverse event or reaction to the IEC in the specified format within 10 working
- For industry sponsored research, trials of drugs or device, sponsors are required to inform investigators of adverse events that occur at other sites. When PI receives such adverse forms from other sites, he/she must notify it to IEC as early as
- All the onsite SAEs will be reviewed by “SAE Committee” which consists of one of the IEC members, legal expert, and chairperson and member secretary. Committee will give it’s detailed remarks on causal relation, compensation, risk benefits assessment
- Final opinion regarding onsite SAEs will be sent to the licensing authority, duly signed by the chairperson within 21 calendar
- Follow-up
- IEC will follow up all studies which are cleared by
- Investigators are required to submit a 6 monthly report to IEC apart from the final
- Any AE, SAE or Suspected Unexpected Serious Adverse Reaction (SUSAR) should be reported to IEC within 24 hours of the
- The IEC reserves the right to review the study or inspect the study site during the study period. The decision can be continuation, suspension or termination of
- In case of premature suspension/termination of study, the applicant must notify the IEC of the reasons for suspension/termination.
- Financial Matters for IEC For projects within the college (GDCH, Nagpur), no fee will be
The committee may, after discussion with the Dean of the institute, decide upon/ announce/ change/amend/ alter, from time to time, announce and implement the fees for Clinical Trial / Study /Protocol / Research Proposal. The said fees are payable by the Sponsors. The fees chargeable may be classified variously under heads such as: protocol processing fees, per amendment/follow-up study fees, and expedited review fees.
The processing fees are to be paid before approval of the protocol/amendment. An IEC approval letter will not be
issued until copy of the receipt of processing fee is submitted to the Member Secretary.
There will no additional fee charged for the review and approval of amended protocol and consent version.
Fees charged for various types of documents.
- Review of New research proposal 30000/-
- Major amendment to a Sponsor based and IEC approved project (initiated by Sponsor) 10,000/-
- Minor amendment to a sponsor-based and IEC approved project 5000/-
- Mode of Payments
Fees/Payments shall be paid by Cheque/DD drawn in favour of
Pan ID:
As per ICMR- Guidelines for vulnerable population (page no 28 ICMR- Guidelines 2006) vulnerable groups: Effort may be made to ensure that individuals or communities invited for research be selected in such a way that the burdens and benefits of the research are equally distributed.
- Research on genetics should not lead to racial inequalities;
- Persons who are economically or socially disadvantaged should not be used to benefit those who are better off than them;
- Rights and welfare of mentally challenged and mentally differently able persons who are incapable of giving informed consent or those with behavioural disorders must be protected. Appropriate proxy consent from the legal guardian should be taken after the person is well informed about the study, need for participation, risks and benefits involved and the privacy and confidentiality procedures. The entire consent process should be properly documented;
- Adequate justification is required for the involvement of participants such as prisoners, students, subordinates, employs, service personnel etc. who have reduced autonomy as research participants, since the consent provided may be under duress or various other compelling
Abbreviations
-ANDA : Abbreviated New Drug Application
- BA : Bioavailability
- BE : Bioequivalence
- CDER : Centre for Drug Evaluation and Research
- CDSCO : Central Drug Standard Control Organization India
- CFR : Code of Federal Regulation, a Publication of US FDA
- CIOMS : Council for International Organization of Medical Sciences
- COA : Certificate of Analysis
- COMP : For the Designation of “Orphan” Medicines for rare diseases (See EMEA)
- CPMP : Term Responsible for Medicines for Human Use (See EMEA)
- CRF : Case Record Form
- CSR : Clinical Study Report
- CTA : Certificate of Authorization
- CTC : Clinical Trial Certificate (Not in vogue due to introduction of CTA)
- CTX : Certificate of Exemption (Not in vogue due to introduction of CTA)
- CVMP : For Veterinary Medicines (See EMEA)
- DCG : Drug Controller General
- DCGI : Drug Controller General of India
- DOH : Director of Health
- DTAB : Drugs Technical Advisory Board
- EC : Ethics Committee
- EMEA : The European Agency for Evaluation of Medical Products, a decentralized body of the European union (EU) with headquarters in London. It has three committees, namely CPMP, CVMP and
- FDA : U.S. Food and Drug Administration
- GCP : Good Clinical Practice
- GEAC : Genetic Engineering Approval Committee
- GLP : Good Laboratory Practice
- GMP : Good Manufacturing Practice- HHS : Health and Human Service
- IB : Investigator’s Brochure
- ICD : Informed Consent Document (=ICF : Informed Consent Form)
- ICH : International Conference on Harmonization
- ICMR : Indian Council of Medical Research
- IDE : Investigational Device Exemption
- IEC : Independent Ethics Committee
- IEC : Institutional Ethics Committee
- IND : Investigational New Drug
- IRB : Institutional Review Board. The FDA term for Ethics Committee.
- MHRA : Medicines and Health Products Regulatory Agency (U.K.) (Advised by CSM)
- NAI : No Objectionable Conditions or Practices were found during
- HMSC : Human Mesenchymal stem
- MOU: Memorandum of
Appendix I: Format for approval of Institutional Ethics Committee
Dr.
Dear, Dr. Ref: your letter dated
Principal Investigator
The Institutional Ethics Committee reviewed and discussed your application to review the Protocol/amendment/ ICF entitled
‘
’ IEC has reviewed and approved in principle the above mentioned Protocol/ amendment/ ICF The following below study-related documents have been reviewed in the meeting:
Sr.No: | Submission Documents (For investigational site EC) | Version(s)/Date of document |
The following members of the Institutional Ethics Committee were present at the meeting held on at time A.M/P.M at Government Dental College & Hospital, Nagpur.
Names | Qualification | Affiliations | IEC Designation & Role | Gender |
Please note that the Principal Investigator was invited to explain the protocol. He/She and/ or other study staff members did not participate in the decision making / voting procedures.
Please note that this is “Approval in Principle “The study can not be initiated unless final approval is issued. The Final approval will be issued after completing all the pending items/ documents as per regulatory requirements and IEC SOP’s.
The Institutional Ethics Committee, Government Dental College & Hospital, Nagpur(IECGDCHN) follows procedures that are in compliance with the requirements of ICH (International Conference on Harmonization) guidance related to GCP (Good Clinical Practice), schedule Y and all applicable Indian regulations.
The IECGDCHN expects to be informed about the progress of the study mention frequency as per EC SOP, any SAE occurring in the course of the study, any changes in the protocol and patient information/informed consent and asks to be provided a copy of the final report.
Yours sincerely, Date of Issue-
Member Secretary IECGDCHN
Appendix II – Invitation from the Dean to be a Chairman/Member Secretary/Member of Institutional Ethics Committee (Human studies)
To
Dear Sir / Madam,
On behalf of Government Dental College & Hospital,Nagpur, I request your concurrence for possible appointment as a Chairman/Member Secretary/Member of Institutional Ethics Committee of the institute. Kindly send your written acceptance in the enclosed format and provide short curriculum vitae (C.V) along with attested copies of educational qualifications and experience certificates along with the acceptance letter.
On receipt of your acceptance, I shall send you the formal appointment letter. Thanking you.
DEAN
GDCH,Nagpur
Institutional Ethics Committee
(Human Studies) of Government Dental College & Hospital, Nagpur 2021-2024
Appendix III- Consent to be a Chairman/Member Secretary/Member of Institutional Ethics Committee (Human Studies)
To,
The Dean
Government Dental College & Hospital, Nagpur 440003
Ref: Your Letter No: dated ………/…..…/……………
Respected Sir/Madam,
In response to your letter stated above, I give my consent to become a Chairman/Member Secretary/Member of Institutional Ethics Committee of Maharashtra University of Health Sciences, Nashik. I shall regularly participate in the IEC meeting to review and give my unbiased opinion regarding the ethical issues. I shall be willing for my name, profession and affiliation to be published.
I shall not keep any literature or study related document with me after the discussion and final review.
I shall maintain all the research project related information confidential and shall not reveal the same to anyone other than project related personnel.
I herewith enclose my CV and required document copies. Thanking you,
Date- Yours sincerely
Name of the Chairman / Member Secretary/Member Address & Contact No
Email ID-
Appendix IV: Proposal format for submission of Research Proposal
Name of College | |
Department | |
Name of Primary Investigator | |
Name of Guide |
To,
Member Secretary,
Institutional Ethics Committee, GDCH, Nagpur. Subject: Submission of Research Proposal titled
Dear Sir,
I, , am planning to conduct a research study and thereby enclosing the subject research proposal for your review. If you need any clarification on the same, I shall be available at the meeting convened by your committee to discuss the same.
I am herewith submitting title & Synopsis of my research protocol titled-
Kindly accept the same for IEC approval.
Dr.
(Name & Signature of PI)
Dr. Dr.
(Name & Signature of Guide) (Name & Signature of HoD)
|
Appendix IV: Proposal format for submission of Research Proposal (Checklist) The proposal submitted herewith comprises of the following documents-
Name & Signature of PI- Dt-
Appendix V- CERTIFICATE OF APPROVAL OF RESEARCH PROPOSAL-
(Or as per Certificate required by MUHS,Nashik or any other Recognised Body)
Research Project/ Protocol no: Date:
This is to certify that the research project/proposal no
…………………… entitled “………………………………….. ”
submitted by ………………… has been approved by the
Institutional Ethics Committee (Human Studies) in the
meeting held on …………………………., at – am/pm under the following terms and conditions.
- This approval is valid for three years or the duration of the proposal whichever less
- Any serious adverse event occurring during the course of the study should be reported to the IEC within a period of seven
- A half yearly progress report of the proposal has to be submitted to the IEC for
- Any change in the study procedure/site/investigator should be informed to the Member Secretary IEC,GDCH,Nagpur 2021-2024
Appendix VI: PARTICIPANT CONSENT FORM
Date:
Participant’s name:
Address:
Title of the project:
The details of the study have been provided to me in writing and explained to me in my own language. I confirm that I have understood the above study and had the opportunity to ask questions. I understand that my participation in the study is voluntary and that I am free to withdraw at any time, without giving any reason, without the medical care that will normally be provided by the hospital being affected. I agree not to restrict the use of any data or results that arise from this study provided such a use is only for scientific purpose(s). I have been given an information sheet giving details of the study. I fully consent to participate in the above study.
Name & Signature of the participant: Date:
Name & Signature of the witness: Date:
Name & Signature of the investigator: Date:
Appendix VII: CONSENT FORM (in Marathi)
सहभाग संमती फॉमम दिनांक-
सहभाग घेणा–र् ााम व्यक्तीचे नाव :
पत्ता :
प्रकल्पाचे शीममक:
र् ाा अभ्यासाचा(उपक्रमाचा) तपशील मला लेखी स्वरुपांत दप्रान के लेला आहे आिण मला तो स्वतःच्या भार् ाे त स्पष्ट करुन दिलेला
आहे. मी हमी (खात्री) दााे तो / दााे ते की मला वरील उपक्रम /अभ्यास कळालेले आहे आिण मला प्रश्न िवचारण्याची संधी होती. मला
मािहत आहे की माझा र् ाा अभ्यासातील सहभाग ऎच्छिक आहे आिण मी कोणत्याही क्षणी (वेळी) माघार घेण्यासाठी, कोणतेरी कारण न दााे ता मुक्त आहे. मात्र त्यामुळे मला िमळणाऱ्र् ाा कोणत्याही वॆद्यकीर् दमतीवर जी की सवमसाधारणपणे एका
रुग्णलर् ाात दिली जातेत्यावर पररणाम होणार नाही. र् ाा अभ्यासातून उत्तपन होणारी मािहती व पररणाम जो पममत फक्त वॆज्ञािनकदृष्टर् ाा वापरली जाणार असेल तो पममत मी त्यास प्रितबंध न करण्यास सहमत आहे. वरील अभ्यासाची मािहती दााे णारे एक मािहती पत्रक मला दााे ण्यात आलेले आहे. र् ाा अभ्यासासाठी सहभागी होण्याची माझी संपूणम समंती आहे.
सहभागी व्यक्तीची स्वाक्षरी: दिनांक- साक्षीदाााराची स्वाक्षरी : दिनांक-
तपासनीसचीं स्वाक्षरी: दिनांक-
Appendix VII: CONSENT FORM (in Hindi)
प्रितभागी सहमित फॉमम तारीख: प्रितभागी का नाम: पता:
पररर् ाोजना का शीममक: अध्यनम के िववरण िलिखत रूप में मेरे
िलए दप्रान िकर् ाा गर् ाा है और मेरी खुदा की भार् ाा में मुझे
समझार् ाा। मैं इस बात की पुिष्ट है िक मैं ऊपर अध्यनम समझा और सवाल पूछने का अवसर िमला है। मैं समझता हूँ िक इस अध्यनम में मेरी भागीदााारी स्वैच्छिक है और मैं िचिकत्सा दााे खभाल िक सामान्य रूप से प्रभािवत िकर् ाा जा रहा अस्पताल द्वारा दप्रान
िकर् ाा जाएगा िबना, िबना कोई कारण बताए िकसी भी समर् वापस लेने के िलए, स्वतंत्र हूँ। मैं दप्रान की एक ऐसी उपर् ाोग के वल वैज्ञािनक उद्दे श्य (ओं) के िलए है िकसी भी डेटा र् ाा पररणाम है
िक इस अध्यनम से उत्पन्न होने के उपर् ाोग को प्रितबंिधत करने के
िलए सहमत नहीं है। मैं अध्यनम की जानकारी दााे ने के िलए एक सूचना पत्र दिर् ाा गर् ाा है। मैं पूरी तरह से ऊपर अध्यनम में भाग लेने के िलए सहमित दााे ते हैं। भागीदााार के हस्ताक्षर: दिनांक: गवाह के हस्ताक्षर: दिनांक: अन्वेकम के हस्ताक्षर: दिनांक:
GOVT. DENTAL COLLEGE & HOSPITAL, NAGPUR.
INSTITUTIONAL ETHICS COMMITTEE GOVERNMENT DENTAL COLLEGE AND HOSPITAL, NAGPUR
(IEC—GDCHN)
STANDARD OPERATING PROCEDURES (SOPs)
Dr Darshan Dakshindas
STANDARD OPERATING PROCEDURES (SOPs) FOR INSTITUTIONAL ETHICS COMMITTEE (IEC) GOVERNMENT DENTAL COLLEGE & HOSPITAL, NAGPUR (IEC-GDCHN)
EFFECTIVE DATE: 01.01.2021 NEXT REVIEW DATE: 31.12.2024
The location and business address of the committee: Institutional Ethics Committee, GDCH, Nagpur
Room No-20 Ground Floor
Government Dental College & Hospital, GMC Campus, Medical Square, Nagpur-Maharashtra, India Pin- 440003
Phone No- 0712-2744496
Standard Operating Procedures for Institutional Ethics Committee, GDCHN (IEC GDCHN) is a content of Compendium on scientific research and publications at GDCH, Nagpur.
- Introduction: Standard Operating Procedures (SOPs) ICH-GCP guideline defines SOPs as “Detailed, written instructions to achieve uniformity of the performance of a specific function”. SOPs provide the essential link between the guideline on one hand and the actual practice on the other. It is but natural therefore that, all the individual participants, performing their primarily required duty or a specific job and belonging to each and every category of the stakeholder has not only to have a well-defined SOPs but also to observe/follow it very carefully. It goes without saying, therefore, that each stakeholder has to have separate SOPs and IEC is no exception to the rule.
SOPs is for the methodical functioning of any important work to be undertaken, a proper, stepwise, work procedure is necessary. In general, in any SOPs the steps given should be reproducible, e.g. in the case of clinical trials, it will be neither proper nor acceptable to have SOPs that can be applied to just one or specific clinical trial. Broadly speaking SOPs can be in four different areas covering (i) organization of study in general, (ii) prior to study, (iii) Actual or during, and (iv) End of the study.
The SOPs should have the following general outline:
- Must have a number with title or checklist. A set SOPs need not have a checklist, but if included it should be sub- numbered, incorporating the corresponding SOPs
- Reference, if any, to other related
- Person/Personnel i.e. who carried out / who all carry out the
- When and how the procedure is carried out (Note: These are especially required for other stakeholders in clinical trials, as compared to the )
- Date of version in use; date of revision/ amendment/ replacement/ automatically replacing the previous version / name of the person or body responsible for the
- The process of review and revision of SOPs should usually be a regularly done exercise, say every few months. A team should preferably do it. The old version should be archived after approval and implementation of the revised version. This is necessary because Regulatory Authorities may want to know some relevant answer to the question that might
Some benefits of SOPs are:
- It provides a written record of the process. Processes used by several individuals are applied (more) consistently. Team member confidence is increased and performance is enhanced. It helps with the training of new
- Reduces supervisory time/effort. However, these can also be generally applicable to other
- As far as this IEC-GDCHN related Work Document is concerned, a properly constituted IEC, functioning regularly and following its own Standard Operating Procedures is a must. Then alone the studies approved by the IEC stand a good chance of the global acceptability of the outcome of their quality
- Objectives
The objective of Standard Operating Procedures (SOPs) is to ensure quality and consistency in review of clinical research proposals and to follow the ICMR and national ethical guidelines for biomedical research on human subjects.
- Institutional Ethics Committee (IEC)
- Composition of Institutional Ethics Committee
ECs should be multi-disciplinary and multi-sectoral. There should be adequate representation of age and gender.
Preferably 50% of the members should be non-affiliated or from outside the institution. The number of members in an EC should preferably be between 07 and 15 and a minimum of five members should be present to meet the QUORUM requirements. The EC should have a balance between medical and non-medical members/technical and non- technical members, depending upon the needs of the institution.
Generally, the representation on the IEC shall be:
- Chairperson (outside Institution-Nominated by the Dean)
- Member Secretary from Institute
- One basic medical scientist (preferably one Pharmacologist)
- One Clinician
- One legal expert or retired Judge
- One social scientist/ representative of non-governmental organization/ Philosopher/ Ethicist/ Theologian or a similar person
- One lay person from the
- Membership requirements:
- Chairperson-(Non-affiliated)-A well-respected person from any background with prior experience of having served/ serving in an
- Member Secretary-(Affiliated)-
- Should be a staff member of the institution
- Should have knowledge and experience in clinical research and ethics and be motivated and have good communication skills
- Should be able to devote adequate time to this activity which should be protected by the institution
- Basic Medical Scientist(s)- (Affiliated/ Non-Affiliated)-
- In case of EC reviewing clinical trials with drugs, the basic medical scientist should preferably be a pharmacologist
- Clinician(s)- (Affiliated/ Non-Affiliated)
- Should be individual/s with recognized medical qualification, expertise and training
- Legal expert/s- (Affiliated/ Non-Affiliated)-
- Should have a basic degree in Law from a recognized university, with experience
- Desirable: Training in medical
- Social scientist/ philosopher/ ethicist/theologian (Affiliated/ Non-Affiliated)-
- Should be an individual with social/behavioural science/ philosophy/ religious qualification and training and/or expertise and be sensitive to local cultural and moral values. Can be from an NGO involved in health-related activities
- Lay person(s)- (Non-affiliated)
- Literate person from the public or community
- Has not pursued a medical science/Health related career in the last 5 years
- May be a representative of the community from which the participants are to be drawn
- Is aware of the local language, cultural and moral values of the community
- Desirable: involved in social and community welfare activities
-Member/s should be sufficiently qualified through the experience and expertise and sensitive to such issues as community attitudes, to promote respect for its advice and counsel in safeguarding the rights and welfare of human subjects.
-In addition to possessing the professional competence necessary to review the specific research activities, the member/s should be able to ascertain the acceptability of proposed research in terms of institutional commitments and regulations, applicable law, and standards or professional conduct and practice. (Ref 21 CFR part 56.107)
-A member of IEC can be a part of any other IRB/IEC
- Tenure: Membership Duration
- The tenure for Members of the IEC is for a period of Three (03) Years.
- It can be extended to another three (03)
- There will be no bar on the members serving for more than one term but it is desirable to have approximately one third fresh
- A member can be replaced in the event of long-term non- availability (three consecutive meetings). Authority to replace the member shall remain with the Chairman
- Members should maintain confidentiality of all discussions during the meeting and sign a confidentiality form at the start of their term. Each member of the committee will submit a declaration to maintain the confidentiality of the documents submitted to them during their membership period.
- Conflict of interest if any, shall be declared by members of the Institutional Ethical Committee at the beginning of every meeting.
- Resignation/ Replacement of members
To establish polices for removal or Resignation / Replacement of Members Chairman and Member Secretary are responsible for implementing this SOP.
Term of appointment Members of IEC will be appointed for period of 03 years initially which could be extended for another term of 03 years. Extension of membership will be based on the recommendation of the Chairman & Member Secretary of IEC.
Policy for removal of member
- A member may be relieved or terminated of his/her membership in case of conduct not suitable for a member of the Ethics
- Inability to participate in the meetings on any grounds for more than 3 meetings of
- The membership shall be reviewed by the Registrar & chairman, if the member is a regular
- If deemed necessary, the IEC may decide to terminate the membership and recommend to the Chairman IEC for necessary
- In all such situations/circumstances, member secretary will serve a letter of termination to the
- Documentation of the termination will be recorded in the meeting minutes of the next duly constituted IEC meeting and IEC membership circular/roster will be Resignation / Replacement procedure
The members who have resigned may be replaced at the discretion of the appointing authority for the same.
IEC members who decide to resign must provide the Chairman & Member Secretary of IEC the written notification of their proposed resignation date at least 30 calendar days prior to the next scheduled meeting.
In case of resignation, chairman & member secretary would appoint a new member, falling in the same category of membership ex. NGO representative with NGO representative.
- Training of IEC members
- Members should be trained in human research protection, EC functions and SOPs, and should be conversant with ethical guidelines, GCP guidelines (if applicable) and relevant regulations of the
- EC members should undergo initial and continuing training in human research protection, applicable EC SOPs and related regulatory requirements. All trainings should be documented.
- Any change in the relevant guidelines or regulatory requirements should be brought to the attention of all EC members.
- EC members should be aware of local, social and cultural norms and emerging ethical
- IEC/IRB Functions:
Roles and responsibilities of the EC
- The basic responsibility of an EC is to ensure protection of the dignity, rights, safety and well-being of the research participants.
- The EC must ensure ethical conduct of research by the investigator
- The EC is responsible for declaration of conflicts of interest to the Chairperson, if any, at each meeting and ensuring these are recorded in the
- The EC should perform its function through competent initial and continuing review of all scientific, ethical, medical and social aspects of research proposals received by it in an objective, timely and independent manner by attending meetings, participation in discussion and
- The EC must ensure that universal ethics values and international scientific standards are followed in terms of local community values and
- The EC should assist in the development and education of the research community in the given institute (including researchers, clinicians, students and others), responsive to local healthcare
- Responsibilities of members should be clearly defined. The SOPs should be given to EC members at the time of their appointment.
- The EC should ensure that privacy of the individual and confidentiality of data including the documents of EC meetings is
- The EC reviews progress reports, final reports and AE/SAE and gives needful suggestions regarding care of the participants and risk minimization procedures, if
- The EC should recommend appropriate compensation for research related injury, wherever
- The EC should carry out monitoring visits at study sites as and when
- The EC should participate in continuing education activities in research ethics and get updated on relevant guidelines and
- The EC may see that conduct of same/similar research by different investigators from same institution is ‘Me too’ research (replicative) should not to be encouraged and submission of same research to different funding agencies should not be accepted.
- Roles and Responsibilities of IEC members: Chairperson
- Conduct EC meetings, accountable for independent and efficient functioning of the committee
-Ensure active participation of all members (particularly non- affiliated, non-medical/ non- technical) in all discussions and deliberations
-Ratify minutes of the previous meetings
-In case of anticipated absence of Chairperson at a planned meeting, the Chairperson should nominate a committee member as Acting Chairperson or the members present may elect an Acting Chairperson on the day of the meeting. The Acting Chairperson should be a non-affiliated person and will have all the powers of the Chairperson for that meeting.
-Seek COI declaration from members and ensure quorum and fair decision making.
-Handle complaints against researchers, EC members, conflict of interest issues and requests for use of EC data, etc.
Member Secretary-
- Organize an effective and efficient procedure for receiving, preparing, circulating and maintaining each proposal for review.
- Schedule EC meetings, prepare the agenda and minutes
- Organize EC documentation, communication and archiving
- Ensure training of EC secretariat and EC members
- Ensure SOPs are updated as and when required
- Ensure adherence of EC functioning to the SOPs
- Prepare for and respond to audits and inspections
- Ensure completeness of documentation at the time of receipt and timely inclusion in agenda for EC review.
- Assess the need for expedited review/ exemption from
review or full review.
- Assess the need to obtain prior scientific review, invite independent consultant, patient or community representatives.
- Ensure quorum during the meeting and record discussions
and decisions.
Basic Medical Scientist(s)-
- Scientific and ethical review with special emphasis on the intervention, benefit-risk analysis, research design,
methodology and statistics, continuing review process, SAE, protocol deviation, progress and completion report
- For clinical trials, pharmacologist to review the drug safety and
Clinician(s)-
- Scientific review of protocols including review of the intervention, benefit-risk analysis, research design, methodology, sample size, site of study and statistics
- Ongoing review of the protocol (SAE, protocol deviation or violation, progress and completion report)
- Review medical care, facility and appropriateness of the principal investigator, provision for medical car, management and
- Thorough review of protocol, investigators brochure (if applicable) and all other protocol details and submitted documents.
Legal expert/s-
- Ethical review of the proposal, Informed Consent Document (ICD) along with translations, Memorandum of Understanding (MOU), Clinical Trial Agreement (CTA), regulatory approval, insurance document, other site approvals, researcher’s undertaking, protocol specific other permissions, such as, stem cell committee for stem cell research, Human Mesenchymal stem cells (HMSC) for international collaboration, compliance with guidelines etc.
- Interpret and inform EC members about new regulations if any Social scientist/ philosopher/ ethicist/theologian
- Ethical review of the proposal, ICD along with the translations.
- Assess impact on community involvement, socio–cultural context, religious or philosophical context, if any
- Serve as a patient/participant/ societal /community representative and bring in ethical and societal concerns. Lay person(s)-
- Ethical review of the proposal, ICD along with translation(s).
- Evaluate benefits and risks from the participant’s perspective and opine whether benefits justify the risks.
- Serve as a patient/participant/ community representative and bring in ethical and societal concerns.
- Assess on societal aspects if
- Record Keeping and Archiving:
- Curriculum Vitae (CV) of all members of
- Minutes of all meetings duly signed by the Chairperson. Copies of all correspondence with members, researchers and other regulatory
- Copy of existing relevant national and international guidelines on research ethics and laws along with amendments.
- All study related documents (study protocols with enclosed documents, progress reports, and SAEs.) should be archived for minimum of five years after the completion of study. A copy of filled Clinical Record Form (CRF) shall remain with the PI for minimum of ten
- Final report of the completed
- IEC-Operations
- Meeting: Office and Conduct of the Meeting
- The Chairperson will conduct all meetings of the Institutional Ethics Committee. If for reasons beyond control, the Chairperson is not available, an alternate Chairperson will be elected by the members present from among
- The Member Secretary will be responsible for organizing the meetings, maintaining the records and communicating with all concerned. He/she will prepare minutes of the meetings and get them approved by the Chairperson before communicating to members and Principle
- Chairman & member secretary are responsible for implementing this
- The Member Secretary in consultation with the chairman may convene the IEC meeting once in every
- Additional review meeting can also be held with short notice as and when
- All members will receive notification of meeting schedules in
- A minimum of five persons is required to form the quorum without which a decision regarding the research would not be taken. The quorum would have at least one representative from the following group:
- One basic medical scientist (preferably one Pharmacologist)
- One Clinician
- One legal expert or retired Judge
- One social scientist/ representative of non- governmental organization/ Philosopher/ Ethicist/ Theologian or a similar person
- One lay person from the viii Minutes of the IEC meetings, all the proceeding and deliberation will be documented.
Applicant investigator may be invited to present the proposal or elaborate on specific issue.
The Chairperson will conduct all meetings of the Institutional Ethics Committee. If for reasons beyond control, the Chairperson is not available, an alternate Chairperson will be elected by the members present from among themselves.
- Quorum Requirements
A minimum of 5 members including at least one member from the related speciality, in which presentation is due, should be present. All decisions will be taken in the meetings and not by circulation of project proposals. As per revised “Schedule Y’’ of Drugs & Cosmetics Act, 1940, amended in 2005, the ethics committee approving drug trials will have in the quorum at least one representative from the following groups:
- One basic medical scientist (preferably one pharmacologist).
- One clinician
- One legal expert or
- One social scientist
- One lay person from the community
- Independent Consultants:
Institutional Ethics Committee may call upon subject experts as consultants for review of selected research protocols.
These experts may be specialists in ethical or legal aspects, specific diseases or methodologies, or represent specific communities; patient groups or special interest groups e.g. cancer patients, HIV/AIDS positive persons or ethnic minorities. They will not take part in the decision making process and shall have no voting rights.
- Submission of documents for IEC review: Details of documents to be submitted for EC review
- Covering letter to the Member Secretary
- Type of review requested
- Application form for initial review
- The correct version of the informed consent document (ICD) in English and the local language(s). Translation and back translation certificates (if applicable)
- Case record form/questionnaire
- Recruitment procedures: advertisement, notices (if applicable)
- Patient instruction card, diary, etc. (if applicable)
- Investigator’s brochure (as applicable for drug/biologicals/device trials)
- Details of funding agency/sponsor and fund allocation (if applicable)
- Brief curriculum vitae of all the study researchers
- A statement on COI, if any
- GCP training certificate (preferably within 5 years) of investigators (clinical trials)
- Any other research ethics/other training evidence, if applicable as per EC SOP
- List of ongoing research studies undertaken by the principal investigator (if applicable)
- Undertaking with signatures of investigators
- Regulatory permissions (as applicable)
- Relevant administrative approvals (such as HMSC approval for International trials)
- Institutional Committee for Stem Cell Research (IC-SCR) approval (if applicable)
- MoU in case of studies involving collaboration with other institutions (if applicable)
- Clinical trial agreement between the sponsors, investigator and the head of the institution(s) (if applicable).
- Documentation of clinical trial registration (preferable)
- Insurance policy (it is preferable to have the policy and not only the insurance certificate)for study participants indicating conditions of coverage, date of commencement and date of expiry of coverage of risk (if applicable)
- Indemnity policy, clearly indicating the conditions of coverage, date of commencement and date of expiry of coverage of risk (if applicable)
- Any additional document(s), as required by EC (such as other EC clearances for multicentric studies)
- Protocol
- Submission & Review Procedure
1- Researchers should submit research proposals as soft or hard copies to the Member Secretary for review in the prescribed format and required documents as per EC SOPs.
This list is subject to modifications, depending on the type of research, EC SOPs and institutional policies.
- All relevant documents should be enclosed with application.
- For projects within the GDCH,Nagpur, no fee will be charged.
- For trials from outside agencies/sponsors, fee of Rs 30,000 is to be deposited along with application
- The required number of copies of the proposal (14 for ICMR and outside funded projects and for in-house projects as specified) along with the application and documents in prescribed format duly signed by the PI and Co-investigators/ Collaborators should be forwarded by the Head of the Department (Institute).
- The Member Secretary will acknowledge the receipt of proposal and indicate any lacunae. Missing information should be replied within two weeks or as specified
- The date of meeting will be intimated to the PI who should be available to offer clarifications if
- The decision of Institutional Ethics Committee will be communicated in writing. If revision is to be made, the revised document in required number of copies should be submitted within a stipulated period of time as specified in the
- Review of Projects:
- Meetings of Institutional Ethics Committee shall be held at scheduled intervals as prescribed. Additional meetings will be held as and when
- The proposals will be sent to members at least 2 weeks in advance.
- The Member Secretary shall screen the proposals for their completeness and depending on the risk involved categorize them into three types, namely-
Exemption from review, Expedited review, and Full committee review.
- Decisions will be taken by consensus after discussions, and voting will be done if
- Principle Investigator should be available during the meeting and will give brief presentation of his proposal. He may be asked for any
- Independent consultants/Experts may be invited to offer their opinion on specific research
- The decisions of the meeting shall be recorded in the form of minutes and shall be confirmed during the next meeting.
- Element of Review of Proposed Projects:
- Scientific design and conduct of the
- Approval of scientific review committee/Research committee and other regulatory
- Assessment of predictable risks, harms and potential benefits.
- Procedure for selection of subjects including inclusion/exclusion, withdrawal criteria and other issues like sample size and advertisement
- Management of research related injuries, adverse events and compensation
- Justification for placebo in control arm, if
- Availability of products to the trial subjects after the study, if
- Patient information sheet and informed consent form in English/Hindi and local
- Protection of privacy and confidentiality of
- Involvement of the community, wherever
- Protocol and proforma of the study including the consent form.
- Plans for data analysis and
- Adherence to Regulatory requirements and applicable guidelines.
- Competence of Investigators, research and supporting staff.
- Facilities and Review Process:
- A member shall withdraw from the meeting during the decision procedure concerning an application where a conflict of interest arises. This shall be indicated to the chairperson prior to the review of the application and recorded in the minutes.
- Only members will make the decision. The decisions shall be taken in the absence of Investigators, representatives of sponsors and Decision may be of approval, rejection or recommendation to revise the proposals. Specific suggestions for modifications and reasons for rejection should be
- Revised proposals may be subjected to an expedited review.
- All approved proposals will be subject to the laid down standard conditions. Additional conditions may be added by the Institutional Ethics
- Expedited Review: Proposals which are recommended for minor revisions will be reviewed by a sub committee appointed by the Institutional Ethics Committee (IEC) for clearance and approved by the Chairperson. The approvals will be reported in the next Institutional Ethical Committee meeting by Secretary. The revised form of proposals requiring major changes will be reviewed at the next IEC meeting. Rejected proposals may be reconsidered only if a very strong background is there.
- Periodic updates to be submitted to IEC
- All amendments in protocol/ ICF/ IB or any new information available related to IEC approved protocols must be submitted to
- The investigator/Sponsor is responsible to supply ongoing safety updates, progress reports (periodic) of trial status report, re-approvals (if required) to the IRB/IEC.
- PI is required to inform IEC in writing after the site is initiated
- Half yearly progress report to be submitted in the last week of every December and June. PI’s are requested to submit 2 copies of each report to IEC, one to be given back to PI, one for IEC
- After receiving IEC approval, even if site is not initiated half yearly report must be submitted mentioning reasons for not initiating the site
- The P.I. will be required to intimate the name/s designation and nature of work/ responsibilities entrusted to all the members of his/her team in the said protocol. This information will have to be submitted by the P.I. along with the submission letter addressed to the Member Secretary. Change/s in such a job/responsibility profile will be immediately intimated to the Member
- If an assistant whose name is not intimated is entrusted the said job/ responsibility, it will be considered as a breach and the IEC will take appropriate action in the matter and this decision shall be
- The Principal Investigator and the said team shall abide by privacy and confidentiality when the study is approved by the
- Communication of decision of Institutional Ethics Committee
- Decision will be communicated to Principal Investigator (PI) by the Member Secretary in
- Suggestions for modifications and reasons for rejection shall also be communicated to the
- Reporting of Serious Adverse Event (SAE)
When a subject participating in the study develops any SAE , the PI must promptly report the incident to IEC as well as concerned authorities including regulatory bodies if applicable. Following are the rules at GDCH,Nagpur.
- In the case of site related serious adverse event, the Initial SAE report along with copy of SAE form Appendix XI ( as defined by Schedule Y amendment 30 Jan 2013) is to be submitted within 24
- If the adverse event was anticipated in the protocol and the subject was informed about the possibility of event in the Informed Consent Form (ICF) there is no need to inform IEC unless the adverse event was unexpectedly serious, life threatening or
- If the adverse event was unanticipated, unexpectedly serious, life-threatening or fatal, the adverse event must be reported to IEC through Member Secretary within 24
- If the study is being supported by an industry sponsor, the PI is also responsible for notifying the sponsor. The sponsor must notify the regulatory authorities within 24
- If the PI holds the “Investigational New Drug or Device” in his/her name, he/she is required to notify the regulatory authorities of the adverse event within 24 hours in addition to notifying it to
- Notifying to IEC does not relieve the PI from his/her responsibility to notify the sponsor or regulatory
- The PI must submit a written report of the adverse event or reaction to the IEC in the specified format within 10 working
- For industry sponsored research, trials of drugs or device, sponsors are required to inform investigators of adverse events that occur at other sites. When PI receives such adverse forms from other sites, he/she must notify it to IEC as early as
- All the onsite SAEs will be reviewed by “SAE Committee” which consists of one of the IEC members, legal expert, and chairperson and member secretary. Committee will give it’s detailed remarks on causal relation, compensation, risk benefits assessment
- Final opinion regarding onsite SAEs will be sent to the licensing authority, duly signed by the chairperson within 21 calendar
- Follow-up
- IEC will follow up all studies which are cleared by
- Investigators are required to submit a 6 monthly report to IEC apart from the final
- Any AE, SAE or Suspected Unexpected Serious Adverse Reaction (SUSAR) should be reported to IEC within 24 hours of the
- The IEC reserves the right to review the study or inspect the study site during the study period. The decision can be continuation, suspension or termination of
- In case of premature suspension/termination of study, the applicant must notify the IEC of the reasons for suspension/termination.
- Financial Matters for IEC For projects within the college (GDCH, Nagpur), no fee will be
The committee may, after discussion with the Dean of the institute, decide upon/ announce/ change/amend/ alter, from time to time, announce and implement the fees for Clinical Trial / Study /Protocol / Research Proposal. The said fees are payable by the Sponsors. The fees chargeable may be classified variously under heads such as: protocol processing fees, per amendment/follow-up study fees, and expedited review fees.
The processing fees are to be paid before approval of the protocol/amendment. An IEC approval letter will not be
issued until copy of the receipt of processing fee is submitted to the Member Secretary.
There will no additional fee charged for the review and approval of amended protocol and consent version.
Fees charged for various types of documents.
- Review of New research proposal 30000/-
- Major amendment to a Sponsor based and IEC approved project (initiated by Sponsor) 10,000/-
- Minor amendment to a sponsor-based and IEC approved project 5000/-
- Mode of Payments
Fees/Payments shall be paid by Cheque/DD drawn in favour of
Pan ID:
- SOP’s For vulnerable population
As per ICMR- Guidelines for vulnerable population (page no 28 ICMR- Guidelines 2006) vulnerable groups: Effort may be made to ensure that individuals or communities invited for research be selected in such a way that the burdens and benefits of the research are equally distributed.
- Research on genetics should not lead to racial inequalities;
- Persons who are economically or socially disadvantaged should not be used to benefit those who are better off than them;
- Rights and welfare of mentally challenged and mentally differently able persons who are incapable of giving informed consent or those with behavioural disorders must be protected. Appropriate proxy consent from the legal guardian should be taken after the person is well informed about the study, need for participation, risks and benefits involved and the privacy and confidentiality procedures. The entire consent process should be properly documented;
- Adequate justification is required for the involvement of participants such as prisoners, students, subordinates, employs, service personnel etc. who have reduced autonomy as research participants, since the consent provided may be under duress or various other compelling
Abbreviations
-ANDA : Abbreviated New Drug Application
- BA : Bioavailability
- BE : Bioequivalence
- CDER : Centre for Drug Evaluation and Research
- CDSCO : Central Drug Standard Control Organization India
- CFR : Code of Federal Regulation, a Publication of US FDA
- CIOMS : Council for International Organization of Medical Sciences
- COA : Certificate of Analysis
- COMP : For the Designation of “Orphan” Medicines for rare diseases (See EMEA)
- CPMP : Term Responsible for Medicines for Human Use (See EMEA)
- CRF : Case Record Form
- CSR : Clinical Study Report
- CTA : Certificate of Authorization
- CTC : Clinical Trial Certificate (Not in vogue due to introduction of CTA)
- CTX : Certificate of Exemption (Not in vogue due to introduction of CTA)
- CVMP : For Veterinary Medicines (See EMEA)
- DCG : Drug Controller General
- DCGI : Drug Controller General of India
- DOH : Director of Health
- DTAB : Drugs Technical Advisory Board
- EC : Ethics Committee
- EMEA : The European Agency for Evaluation of Medical Products, a decentralized body of the European union (EU) with headquarters in London. It has three committees, namely CPMP, CVMP and
- FDA : U.S. Food and Drug Administration
- GCP : Good Clinical Practice
- GEAC : Genetic Engineering Approval Committee
- GLP : Good Laboratory Practice
- GMP : Good Manufacturing Practice- HHS : Health and Human Service
- IB : Investigator’s Brochure
- ICD : Informed Consent Document (=ICF : Informed Consent Form)
- ICH : International Conference on Harmonization
- ICMR : Indian Council of Medical Research
- IDE : Investigational Device Exemption
- IEC : Independent Ethics Committee
- IEC : Institutional Ethics Committee
- IND : Investigational New Drug
- IRB : Institutional Review Board. The FDA term for Ethics Committee.
- MHRA : Medicines and Health Products Regulatory Agency (U.K.) (Advised by CSM)
- NAI : No Objectionable Conditions or Practices were found during
- HMSC : Human Mesenchymal stem
- MOU: Memorandum of
Appendix I: Format for approval of Institutional Ethics Committee
Dr.
Dear, Dr. Ref: your letter dated
Principal Investigator
The Institutional Ethics Committee reviewed and discussed your application to review the Protocol/amendment/ ICF entitled
‘
’ IEC has reviewed and approved in principle the above mentioned Protocol/ amendment/ ICF The following below study-related documents have been reviewed in the meeting:
Sr.No: | Submission Documents (For investigational site EC) | Version(s)/Date of document |
The following members of the Institutional Ethics Committee were present at the meeting held on at time A.M/P.M at Government Dental College & Hospital, Nagpur.
Names | Qualification | Affiliations | IEC Designation & Role | Gender |
Please note that the Principal Investigator was invited to explain the protocol. He/She and/ or other study staff members did not participate in the decision making / voting procedures.
Please note that this is “Approval in Principle “The study can not be initiated unless final approval is issued. The Final approval will be issued after completing all the pending items/ documents as per regulatory requirements and IEC SOP’s.
The Institutional Ethics Committee, Government Dental College & Hospital, Nagpur(IECGDCHN) follows procedures that are in compliance with the requirements of ICH (International Conference on Harmonization) guidance related to GCP (Good Clinical Practice), schedule Y and all applicable Indian regulations.
The IECGDCHN expects to be informed about the progress of the study mention frequency as per EC SOP, any SAE occurring in the course of the study, any changes in the protocol and patient information/informed consent and asks to be provided a copy of the final report.
Yours sincerely, Date of Issue-
Member Secretary IECGDCHN
Institutional Ethics Committee Government Dental College & Hospital, Nagpur.
(2021-2024)
Appendix II – Invitation from the Dean to be a Chairman/Member Secretary/Member of Institutional Ethics Committee (Human studies)
To
Sub: Constitution of Institutional Ethics Committee (Human studies)
Dear Sir / Madam,
On behalf of Government Dental College & Hospital,Nagpur, I request your concurrence for possible appointment as a Chairman/Member Secretary/Member of Institutional Ethics Committee of the institute. Kindly send your written acceptance in the enclosed format and provide short curriculum vitae (C.V) along with attested copies of educational qualifications and experience certificates along with the acceptance letter.
On receipt of your acceptance, I shall send you the formal appointment letter. Thanking you.
DEAN
GDCH,Nagpur
Institutional Ethics Committee
(Human Studies) of Government Dental College & Hospital, Nagpur 2021-2024
Appendix III- Consent to be a Chairman/Member Secretary/Member of Institutional Ethics Committee (Human Studies)
To,
The Dean
Government Dental College & Hospital, Nagpur 440003
Sub: Consent to be a Chairman/Member Secretary/Member of Institutional Ethics Committee (Human Studies)
Ref: Your Letter No: dated ………/…..…/……………
Respected Sir/Madam,
In response to your letter stated above, I give my consent to become a Chairman/Member Secretary/Member of Institutional Ethics Committee of Maharashtra University of Health Sciences, Nashik. I shall regularly participate in the IEC meeting to review and give my unbiased opinion regarding the ethical issues. I shall be willing for my name, profession and affiliation to be published.
I shall not keep any literature or study related document with me after the discussion and final review.
I shall maintain all the research project related information confidential and shall not reveal the same to anyone other than project related personnel.
I herewith enclose my CV and required document copies. Thanking you,
Date- Yours sincerely
Name of the Chairman / Member Secretary/Member Address & Contact No
Email ID-
Appendix IV: Proposal format for submission of Research Proposal
Name of College | |
Department | |
Name of Primary Investigator | |
Name of Guide |
To,
Member Secretary,
Institutional Ethics Committee, GDCH, Nagpur. Subject: Submission of Research Proposal titled
Dear Sir,
I, , am planning to conduct a research study and thereby enclosing the subject research proposal for your review. If you need any clarification on the same, I shall be available at the meeting convened by your committee to discuss the same.
I am herewith submitting title & Synopsis of my research protocol titled-
Kindly accept the same for IEC approval.
Dr.
(Name & Signature of PI)
Dr. Dr.
(Name & Signature of Guide) (Name & Signature of HoD)
|
Appendix IV: Proposal format for submission of Research Proposal (Checklist) The proposal submitted herewith comprises of the following documents-
Name & Signature of PI- Dt-
Appendix V- CERTIFICATE OF APPROVAL OF RESEARCH PROPOSAL-
(Or as per Certificate required by MUHS,Nashik or any other Recognised Body)
Research Project/ Protocol no: Date:
CERTIFICATE
This is to certify that the research project/proposal no
…………………… entitled “………………………………….. ”
submitted by ………………… has been approved by the
Institutional Ethics Committee (Human Studies) in the
meeting held on …………………………., at – am/pm under the following terms and conditions.
- This approval is valid for three years or the duration of the proposal whichever less
- Any serious adverse event occurring during the course of the study should be reported to the IEC within a period of seven
- A half yearly progress report of the proposal has to be submitted to the IEC for
- Any change in the study procedure/site/investigator should be informed to the Member Secretary IEC,GDCH,Nagpur 2021-2024
Appendix VI: PARTICIPANT CONSENT FORM
Date:
Participant’s name:
Address:
Title of the project:
The details of the study have been provided to me in writing and explained to me in my own language. I confirm that I have understood the above study and had the opportunity to ask questions. I understand that my participation in the study is voluntary and that I am free to withdraw at any time, without giving any reason, without the medical care that will normally be provided by the hospital being affected. I agree not to restrict the use of any data or results that arise from this study provided such a use is only for scientific purpose(s). I have been given an information sheet giving details of the study. I fully consent to participate in the above study.
Name & Signature of the participant: Date:
Name & Signature of the witness: Date:
Name & Signature of the investigator: Date:
Appendix VII: CONSENT FORM (in Marathi)
सहभाग संमती फॉमम दिनांक-
सहभाग घेणा–र् ााम व्यक्तीचे नाव :
पत्ता :
प्रकल्पाचे शीममक:
र् ाा अभ्यासाचा(उपक्रमाचा) तपशील मला लेखी स्वरुपांत दप्रान के लेला आहे आिण मला तो स्वतःच्या भार् ाे त स्पष्ट करुन दिलेला
आहे. मी हमी (खात्री) दााे तो / दााे ते की मला वरील उपक्रम /अभ्यास कळालेले आहे आिण मला प्रश्न िवचारण्याची संधी होती. मला
मािहत आहे की माझा र् ाा अभ्यासातील सहभाग ऎच्छिक आहे आिण मी कोणत्याही क्षणी (वेळी) माघार घेण्यासाठी, कोणतेरी कारण न दााे ता मुक्त आहे. मात्र त्यामुळे मला िमळणाऱ्र् ाा कोणत्याही वॆद्यकीर् दमतीवर जी की सवमसाधारणपणे एका
रुग्णलर् ाात दिली जातेत्यावर पररणाम होणार नाही. र् ाा अभ्यासातून उत्तपन होणारी मािहती व पररणाम जो पममत फक्त वॆज्ञािनकदृष्टर् ाा वापरली जाणार असेल तो पममत मी त्यास प्रितबंध न करण्यास सहमत आहे. वरील अभ्यासाची मािहती दााे णारे एक मािहती पत्रक मला दााे ण्यात आलेले आहे. र् ाा अभ्यासासाठी सहभागी होण्याची माझी संपूणम समंती आहे.
सहभागी व्यक्तीची स्वाक्षरी: दिनांक- साक्षीदाााराची स्वाक्षरी : दिनांक-
तपासनीसचीं स्वाक्षरी: दिनांक-
Appendix VII: CONSENT FORM (in Hindi)
प्रितभागी सहमित फॉमम तारीख: प्रितभागी का नाम: पता:
पररर् ाोजना का शीममक: अध्यनम के िववरण िलिखत रूप में मेरे
िलए दप्रान िकर् ाा गर् ाा है और मेरी खुदा की भार् ाा में मुझे
समझार् ाा। मैं इस बात की पुिष्ट है िक मैं ऊपर अध्यनम समझा और सवाल पूछने का अवसर िमला है। मैं समझता हूँ िक इस अध्यनम में मेरी भागीदााारी स्वैच्छिक है और मैं िचिकत्सा दााे खभाल िक सामान्य रूप से प्रभािवत िकर् ाा जा रहा अस्पताल द्वारा दप्रान
िकर् ाा जाएगा िबना, िबना कोई कारण बताए िकसी भी समर् वापस लेने के िलए, स्वतंत्र हूँ। मैं दप्रान की एक ऐसी उपर् ाोग के वल वैज्ञािनक उद्दे श्य (ओं) के िलए है िकसी भी डेटा र् ाा पररणाम है
िक इस अध्यनम से उत्पन्न होने के उपर् ाोग को प्रितबंिधत करने के
िलए सहमत नहीं है। मैं अध्यनम की जानकारी दााे ने के िलए एक सूचना पत्र दिर् ाा गर् ाा है। मैं पूरी तरह से ऊपर अध्यनम में भाग लेने के िलए सहमित दााे ते हैं। भागीदााार के हस्ताक्षर: दिनांक: गवाह के हस्ताक्षर: दिनांक: अन्वेकम के हस्ताक्षर: दिनांक:
